What we do
How do we harness current technology to make the most impact to modern biology and medicine? This question is as wide as the expertise of our group. We work in an interdisciplinary manner, covering areas from computer science to molecular biology. We like technology, and we are not afraid to explore the unknown. We are a member of UCMR (Umeå centre for microbial research) as well as Icelab (Integrated science lab). We believe the key to success is to collaborate with others, build critical mass, and exploit opportunities.
Can we phenotype with higher precision?
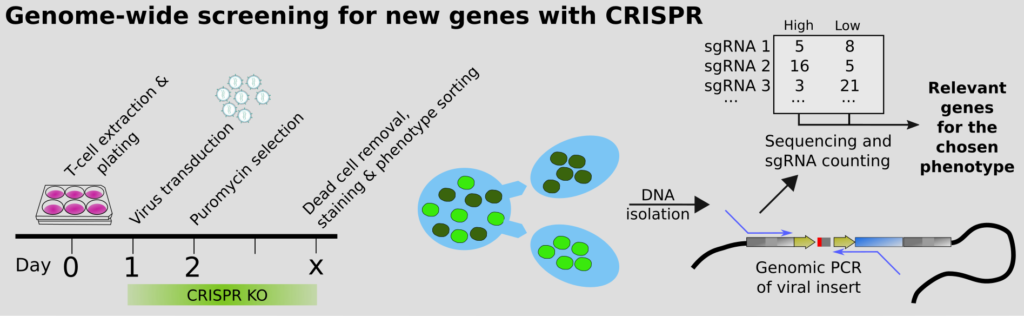
Our end-goal is to help understand all of biology. This can only be achieved by perturbing the system (i.e. knocking out genes) and see what happens. Classical screening for genes that regulate a biological process typically involves multiwell plates, but this is extremely expensive. The modern approach is to instead perform pooled CRISPR screens, which we were among the first to do, in the context of CD4 T cell differentiation; more recently also for the essentiality of genes in Malaria, together with Ellen Bushell. However, how to we guarantee that we get high quality scores for the phenotypes? We have developed an improved approach using molecular inversion probes (MIPs; also called padlocks). These overcome issues with PCR, such as GC%-bias, and enable precise counting of the originating DNA molecules. Statistics for NGS commonly rely on the negative binomial assumption, but we show that it breaks down when you work with a small number of molecules, leading to inflated statistics or simply bizarre conclusions. Complicating the molecular biology a bit thus seems motivated to maximize the value of pooled screens.
Can we improve our understanding of the immune system and improve CAR T cell design using modern single-cell technology?
We have a particular interest in the immune system, especially to improve treatment of cancer. Using single-cell technology, with new readouts, we have especially found a new JUNB+ state of CD4 T cells. This state seems better delineated from an epigenetic standpoint rather than using transcriptomics. It seems intertwined with memoryness, as well as circadian rhythm and cell cycle, which somehow might explain why it is hard to separate. We however also find it in several previous single-cell atlases, and it is especially prevalent in exhausted CAR T cells. A better understanding of this state, which does not conform to the dogma of CD4 T cell subtypes, might thus be key to understanding several properties of T cells, including how to better make them fight cancer.

Our strong side is on the technology, and we are happy to be collaborating with others who contribute more classical biological know-how. This includes for example Isabelle Magalhaes for CAR T cells (T cells genetically modified to target cancer), and Nicole Boucheron for the T cell involvement in allergic asthma. We have also worked with Mattias Forsell on the related B cells, and Anna Överby for the innate immune response during TBE, and Annasara Lenman during SARS-CoV-2.
Can we better distill knowledge out of large datasets?
A huge problem with modern large-scale omics is that we generate huge amounts of data, but aren’t sure how to turn it into working knowledge. But what even is knowledge? We are investigating several routes, all involving machine learning; for example, we have developed a new framework Nando to predict gene expression (i.e. gene regulatory networks), and we have applied it to our T cell atlas. We are also investigating machine learning-based methods of mapping data to our cognitive model through novel latent space formulations and variational autoencoders (VAEs), in collaboration with Tommy Löfstedt. Based on our analysis of the bias of which genes we have uncovered, a constructivist approach to expanding our knowledge is likely motivated.
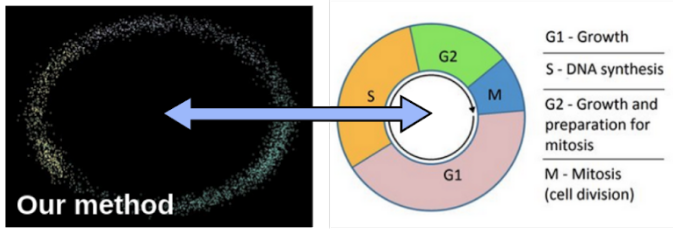
Can predict disease through measuring telomeres?
Our group is investigating how telomeres can be measured at the single-cell level. We envision that this can be used to provide earlier detection of cancer, as especially needed for pancreatic cancer (10% survival, 5 years after diagnosis).
In our first study, we noticed that telomeric fragments are present in ATAC-seq! We first thought that this easily can be used to get an idea about telomere lengths. Unfortunately this signal is too confounded by other factors such as the overall capability of Tn5 the tagment the genome. Tn5 also has sequence biases that complicate the analysis. However, we show that at least telomere fragments can be indicative of overall cell division status and other processes that affect broad chromatin accessibility.
We are now in the process of developing a new tailored single-cell telomere measurement protocol, and results are so far looking promising. Stay tuned!

Can we apply single-cell technology to microbes?
Single-cell technology has so far primarily been used on eukaryotes, because these have turned out to be somewhat simple to approach. Microbes are however at least as important. We are now collaborating especially with Laura Carroll, Kemal Avican and Linas Mažutis, using a new single-cell platform, to be able to also do microbial single-cell analysis. See our paper!

Can we analyze genomic data in new ways?
Most single-cell data is RNA-seq of a single organism. This does not work well for mixes of species, such as microbial genomes. To overcome this, we developed the Bascet/Zorn pipeline, implemented in R and Rust for performance. Additionally, we have innovated in reference-free data comparison by comparing them in high-dimensional randomly projected spaces. These techniques, which also power customer product recommendations at big websites, can now also be applied to genomes!

The future language of bioinformatics: Rust? 🦀
R and Python are not suitable for handling large amounts of data. In some cases, Python can solve problems on GPUs using, e.g., PyTorch, but far from all tasks fit into this framework. Our group is standardizing on using Rust for our demanding calculations. This language provides safety and high productivity through static typing (using recent innovations from research languages such as Haskell), and speed through low-level memory management as in C++. We argue that basic Rust programming is no harder than Java, but that the language is better suited for demanding tasks and enables aggressive optimization of key tasks without the need to mix or switch languages.
Rust is also great for front-end web development, and we have effectively kicked out Javascript/React. See e.g. https://flexo.microbe.dev/, https://sismis.microbe.dev/ and https://www.btyper.app/ for some examples (collaboration with Laura Carroll).
Our group is especially interested in students and bioinformaticians who aren’t afraid to roll up their sleeves and code some Rust! 🦀